|
Literature for DOID 2843: long QT syndrome
Xenbase Articles
(
| Functional and clinical characterization of a mutation in KCNJ2 associated with Andersen-Tawil syndrome., Lu CW,Lin JH,Rajawat YS,Jerng H,Rami TG,Sanchez X,DeFreitas G,Carabello B,DeMayo F,Kearney DL,Miller G,Li H,Pfaffinger PJ,Bowles NE,Khoury DS,Towbin JA, J Med Genet. August 1, 2006; 43(8):1468-6244. |
| The MinK-related peptides., McCrossan ZA,Abbott GW, Neuropharmacology. November 1, 2004; 47(6):1873-7064. |
| Heteromerization of Kir2.x potassium channels contributes to the phenotype of Andersen's syndrome., Preisig-Müller R,Schlichthörl G,Goerge T,Heinen S,Brüggemann A,Rajan S,Derst C,Veh RW,Daut J, Proc Natl Acad Sci U S A. May 28, 2002; 99(11):1091-6490. |
| Novel characteristics of a misprocessed mutant HERG channel linked to hereditary long QT syndrome., Ficker E,Thomas D,Viswanathan PC,Dennis AT,Priori SG,Napolitano C,Memmi M,Wible BA,Kaufman ES,Iyengar S,Schwartz PJ,Rudy Y,Brown AM, Am J Physiol Heart Circ Physiol. October 1, 2000; 279(4):0363-6135. |
| Stilbenes and fenamates rescue the loss of I(KS) channel function induced by an LQT5 mutation and other IsK mutants., Abitbol I,Peretz A,Lerche C,Busch AE,Attali B, EMBO J. August 2, 1999; 18(15):0261-4189. |
| Characterization of a voltage-gated K+ channel beta subunit expressed in human heart., England SK,Uebele VN,Shear H,Kodali J,Bennett PB,Tamkun MM, Proc Natl Acad Sci U S A. July 3, 1995; 92(14):1091-6490. |
| Mechanistic basis for the pathogenesis of long QT syndrome associated with a common splicing mutation in KCNQ1 gene., Tsuji K,Akao M,Ishii TM,Ohno S,Makiyama T,Takenaka K,Doi T,Haruna Y,Yoshida H,Nakashima T,Kita T,Horie M, J Mol Cell Cardiol. March 1, 2007; 42(3):1095-8584. |
| Biophysical properties of zebrafish ether-à-go-go related gene potassium channels., Scholz EP,Niemer N,Hassel D,Zitron E,Bürgers HF,Bloehs R,Seyler C,Scherer D,Thomas D,Kathöfer S,Katus HA,Rottbauer WA,Karle CA, Biochem Biophys Res Commun. April 3, 2009; 381(2):1090-2104. |
|
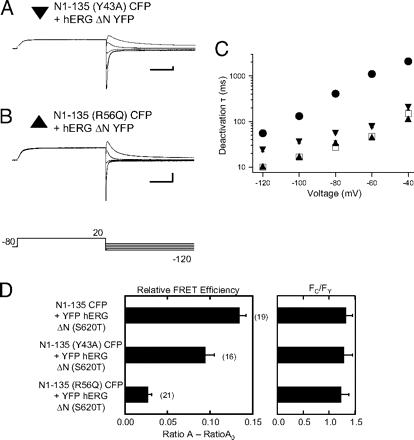
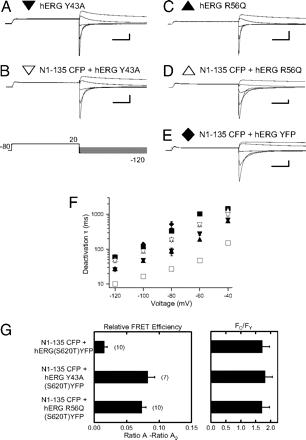
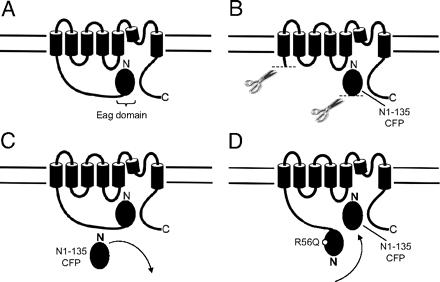
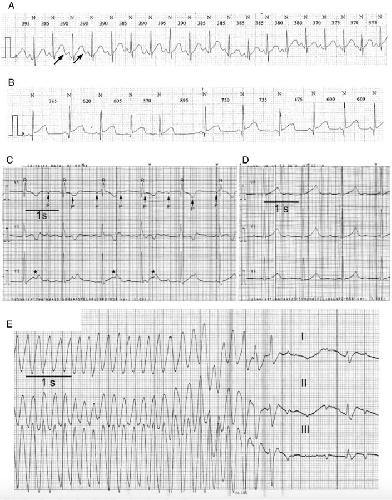
A recombinant N-terminal domain fully restores deactivation gating in N-truncated and long QT syndrome mutant hERG potassium channels.,
Gustina AS,Trudeau MC,
Proc Natl Acad Sci U S A. August 4, 2009; 106(31):1091-6490.
|
| Biophysical properties of mutant KCNQ1 S277L channels linked to hereditary long QT syndrome with phenotypic variability., Aidery P,Kisselbach J,Schweizer PA,Becker R,Katus HA,Thomas D, Biochim Biophys Acta. April 1, 2011; 1812(4):0006-3002. |
| Identification and functional characterization of KCNQ1 mutations around the exon 7-intron 7 junction affecting the splicing process., Tsuji-Wakisaka K,Akao M,Ishii TM,Ashihara T,Makiyama T,Ohno S,Toyoda F,Dochi K,Matsuura H,Horie M, Biochim Biophys Acta. November 1, 2011; 1812(11):0006-3002. |
| Structural basis of slow activation gating in the cardiac I Ks channel complex., Strutz-Seebohm N,Pusch M,Wolf S,Stoll R,Tapken D,Gerwert K,Attali B,Seebohm G, Cell Physiol Biochem. January 1, 2011; 27(5):1421-9778. |
|
Impaired ion channel function related to a common KCNQ1 mutation - implications for risk stratification in long QT syndrome 1.,
Aidery P,Kisselbach J,Schweizer PA,Becker R,Katus HA,Thomas D,
Gene. December 10, 2012; 511(1):1879-0038.
|
|
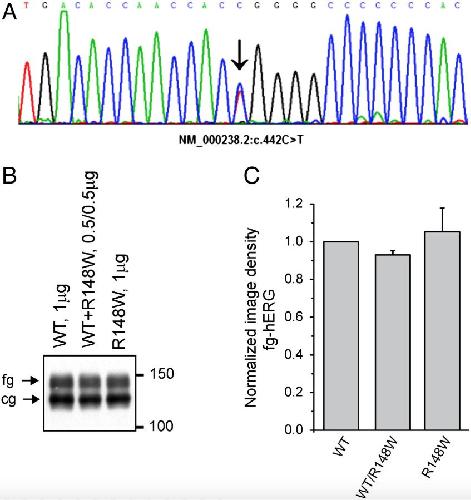
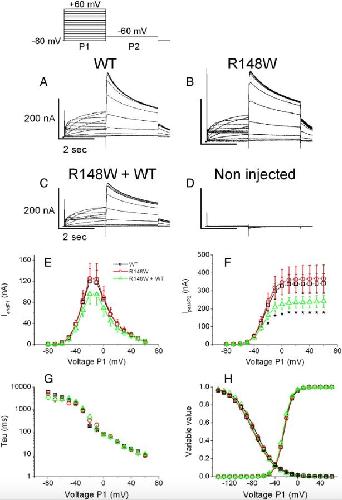
The variant hERG/R148W associated with LQTS is a mutation that reduces current density on co-expression with the WT.,
Mechakra A,Vincent Y,Chevalier P,Millat G,Ficker E,Jastrzebski M,Poulin H,Pouliot V,Chahine M,Christé G,
Gene. February 25, 2014; 536(2):1879-0038.
|
| Moving gating charges through the gating pore in a Kv channel voltage sensor., Lacroix JJ,Hyde HC,Campos FV,Bezanilla F, Proc Natl Acad Sci U S A. May 13, 2014; 111(19):1091-6490. |
|
High incidence of functional ion-channel abnormalities in a consecutive Long QT cohort with novel missense genetic variants of unknown significance.,
Steffensen AB,Refaat MM,David JP,Mujezinovic A,Calloe K,Wojciak J,Nussbaum RL,Scheinman MM,Schmitt N,
Sci Rep. January 12, 2015; 5:2045-2322.
|
| Iron Overload Leading to Torsades de Pointes in β-Thalassemia and Long QT Syndrome., Refaat MM,El Hage L,Steffensen AB,Hotait M,Schmitt N,Scheinman M,Badhwar N, Card Electrophysiol Clin. March 1, 2016; 8(1):1877-9190. |
| Functional and clinical characterization of KCNJ2 mutations associated with LQT7 (Andersen syndrome)., Tristani-Firouzi M,Jensen JL,Donaldson MR,Sansone V,Meola G,Hahn A,Bendahhou S,Kwiecinski H,Fidzianska A,Plaster N,Fu YH,Ptacek LJ,Tawil R, J Clin Invest. August 1, 2002; 110(3):1558-8238. |
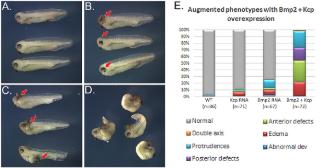
| Modeling human craniofacial disorders in Xenopus., Dubey A,Saint-Jeannet JP, Curr Pathobiol Rep. March 1, 2017; 5(1):2167-485X. |
| Effects of trimethoprim-sulfadiazine and detomidine on the function of equine Kv 11.1 channels in a two-electrode voltage-clamp (TEVC) oocyte model., Trachsel DS,Tejada MA,Groesfjeld Christensen V,Pedersen PJ,Kanters JK,Buhl R,Calloe K,Klaerke DA, J Vet Pharmacol Ther. August 1, 2018; 41(4):1365-2885. |
| Unexplained cardiac arrest: a tale of conflicting interpretations of KCNQ1 genetic test results., Chua HC,Servatius H,Asatryan B,Schaller A,Rieubland C,Noti F,Seiler J,Roten L,Baldinger SH,Tanner H,Fuhrer J,Haeberlin A,Lam A,Pless SA,Medeiros-Domingo A, Clin Res Cardiol. August 1, 2018; 107(8):1861-0692. |
|
ANK2 functionally interacts with KCNH2 aggravating long QT syndrome in a double mutation carrier.,
Gessner G,Runge S,Koenen M,Heinemann SH,Koenen M,Haas J,Meder B,Thomas D,Katus HA,Schweizer PA,
Biochem Biophys Res Commun. May 14, 2019; 512(4):1090-2104.
|
|
Functional characterization of Kv11.1 (hERG) potassium channels split in the voltage-sensing domain.,
de la Peña P,Domínguez P,Barros F,
Pflugers Arch. July 1, 2018; 470(7):1432-2013.
|
|
The molecular determinants of R-roscovitine block of hERG channels.,
Cernuda B,Fernandes CT,Allam SM,Orzillo M,Suppa G,Chia Chang Z,Athanasopoulos D,Buraei Z,
PLoS One. September 3, 2019; 14(9):1932-6203.
|
|
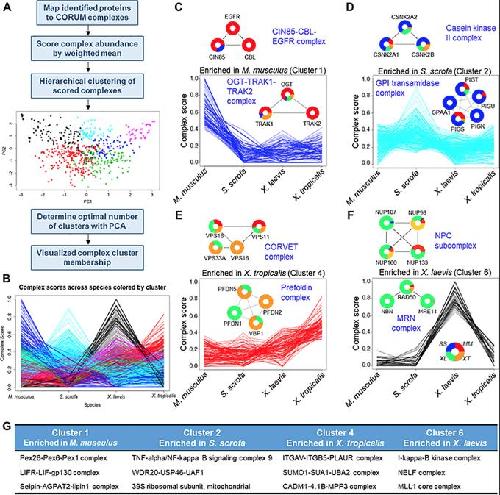
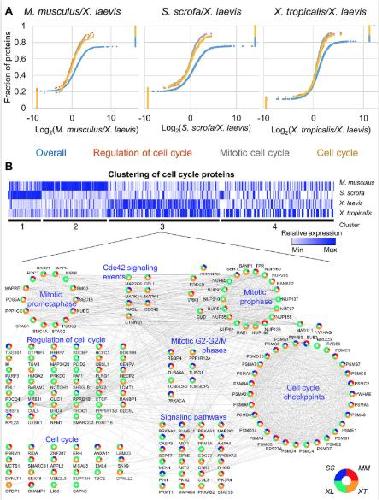
Conservation and divergence of protein pathways in the vertebrate heart.,
Federspiel JD,Tandon P,Wilczewski CM,Wasson L,Herring LE,Venkatesh SS,Cristea IM,Conlon FL,
PLoS Biol. September 6, 2019; 17(9):1545-7885.
|
|
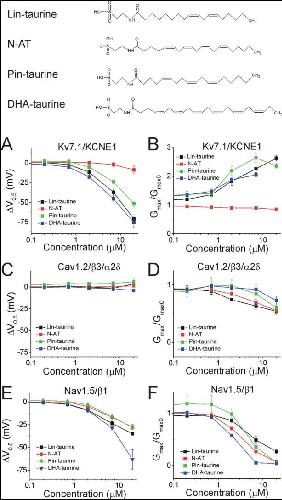
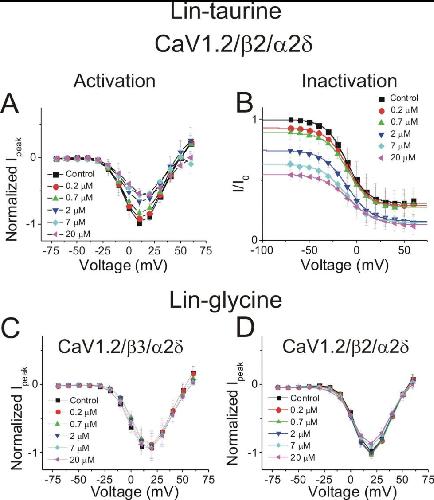
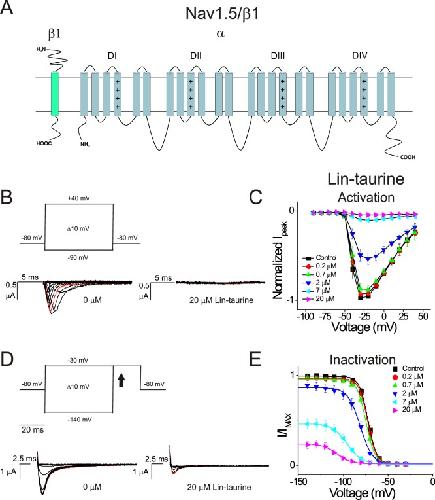
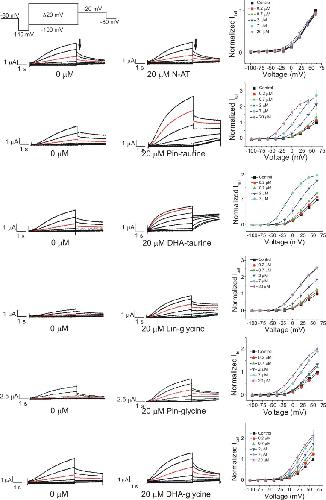
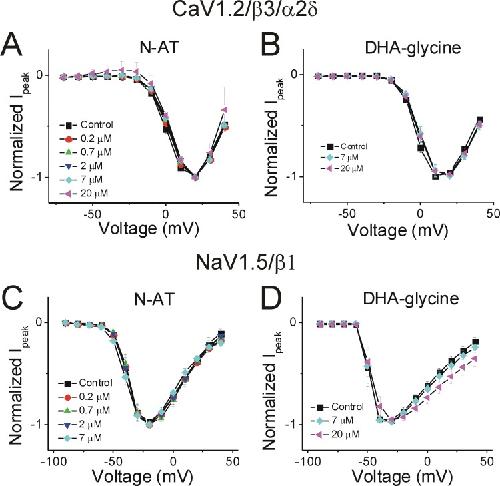
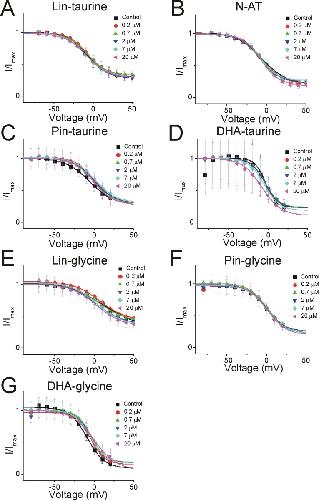
Polyunsaturated fatty acid analogues differentially affect cardiac NaV, CaV, and KV channels through unique mechanisms.,
Bohannon BM,de la Cruz A,Wu X,Jowais JJ,Perez ME,Dykxhoorn DM,Liin SI,Larsson HP,
Elife. March 24, 2020; 9:2050-084X.
|
|
Andersen-Tawil Syndrome Is Associated With Impaired PIP2 Regulation of the Potassium Channel Kir2.1.,
Handklo-Jamal R,Meisel E,Yakubovich D,Vysochek L,Beinart R,Glikson M,McMullen JR,Dascal N,Nof E,Oz S,
Front Pharmacol. April 7, 2020; 11:1663-9812.
|
|
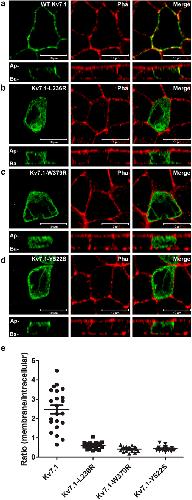
Molecular Mechanism of Autosomal Recessive Long QT-Syndrome 1 without Deafness.,
Oertli A,Rinné S,Moss R,Kääb S,Seemann G,Beckmann BM,Decher N,
Int J Mol Sci. January 23, 2021; 22(3):1422-0067.
|
| Compound Heterozygous KCNQ1 Mutations Causing Recessive Romano-Ward Syndrome: Functional Characterization by Mutant Co-expression., González-Garrido A,Domínguez-Pérez M,Jacobo-Albavera L,López-Ramírez O,Guevara-Chávez JG,Zepeda-García O,Iturralde P,Carnevale A,Villarreal-Molina T, Front Cardiovasc Med. February 22, 2021; 8:2297-055X. |
|
Bioelectric signaling: Reprogrammable circuits underlying embryogenesis, regeneration, and cancer.,
Levin M,
Cell. April 15, 2021; :1097-4172.
|
|
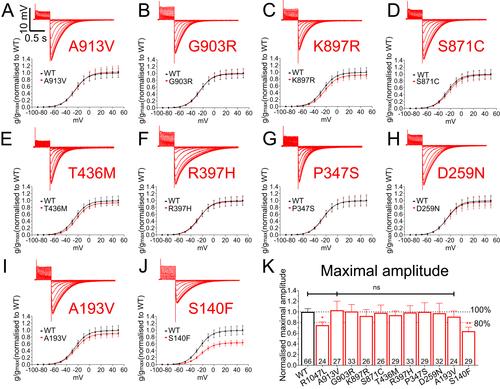
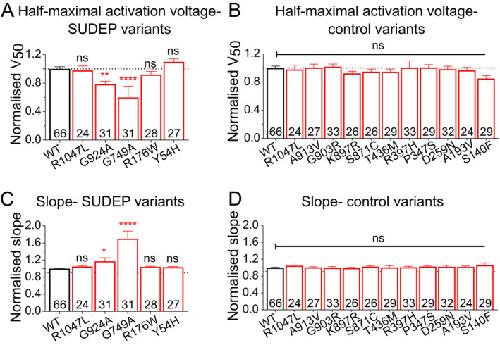
Loss-of-function variants in Kv 11.1 cardiac channels as a biomarker for SUDEP.,
Soh MS,Bagnall RD,Bennett MF,Bleakley LE,Mohamed Syazwan ES,Phillips AM,Chiam MDF,McKenzie CE,Hildebrand M,Crompton D,Bahlo M,Semsarian C,Scheffer IE,Berkovic SF,Reid CA,
Ann Clin Transl Neurol. July 1, 2021; 8(7):2328-9503.
|
|
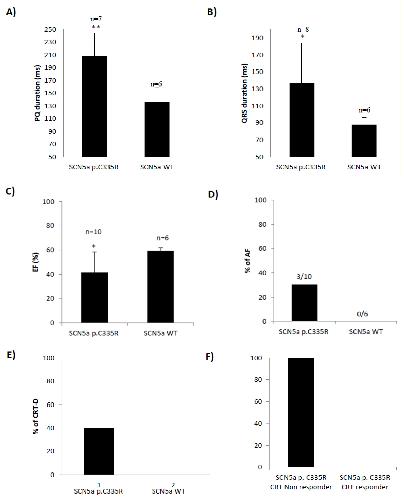
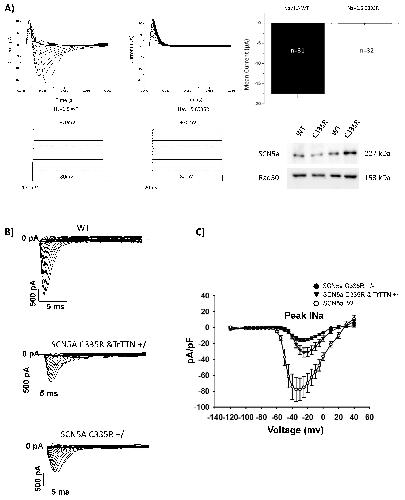
Identification of SCN5a p.C335R Variant in a Large Family with Dilated Cardiomyopathy and Conduction Disease.,
Sedaghat-Hamedani F,Rebs S,El-Battrawy I,Chasan S,Krause T,Haas J,Zhong R,Liao Z,Xu Q,Zhou X,Zhou X,Akin I,Zitron E,Frey N,Streckfuss-Bömeke K,Kayvanpour E,
Int J Mol Sci. November 30, 2021; 22(23):1422-0067.
|
|
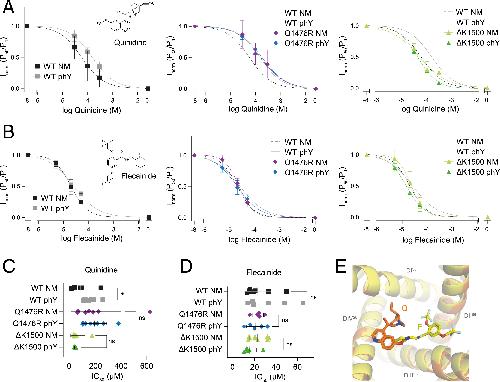
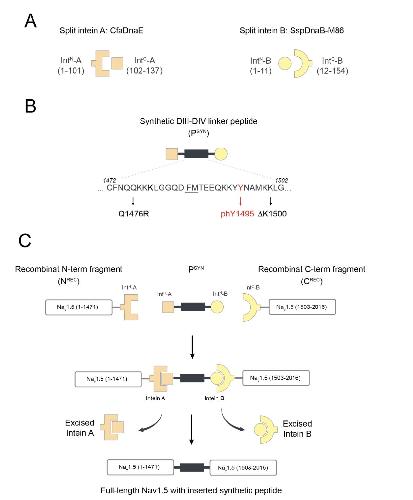
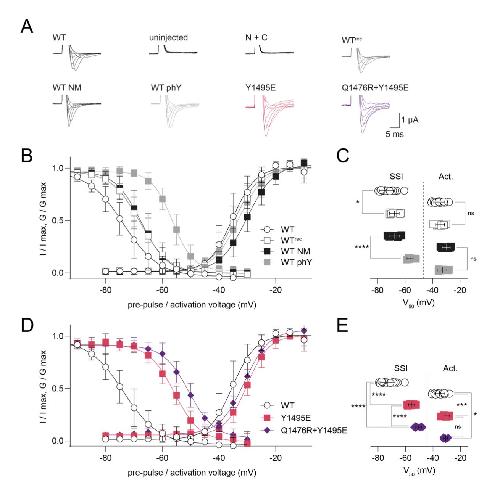
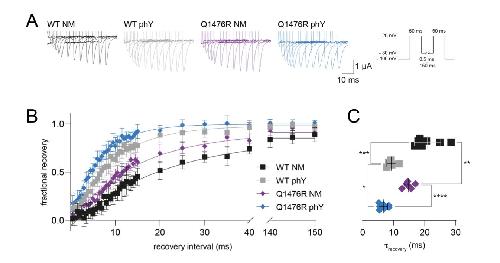
Functional cross-talk between phosphorylation and disease-causing mutations in the cardiac sodium channel Nav1.5.,
Galleano I,Harms H,Choudhury K,Khoo K,Delemotte L,Pless SA,
Proc Natl Acad Sci U S A. August 17, 2021; 118(33):1091-6490.
|
|
Pharmacological rescue of specific long QT variants of KCNQ1/KCNE1 channels.,
Zou X,Wu X,Sampson KJ,Colecraft HM,Larsson HP,Kass RS,
Front Physiol. January 1, 2022; 13:1664-042X.
|
|
Long-QT mutations in KCNE1 modulate the 17β-estradiol response of Kv7.1/KCNE1.,
Erlandsdotter LM,Giammarino L,Halili A,Nikesjö J,Gréen H,Odening KE,Liin SI,
Sci Adv. March 17, 2023; 9(11):2375-2548.
|
|
Functional Characterization of a Spectrum of Novel Romano-Ward Syndrome KCNQ1 Variants.,
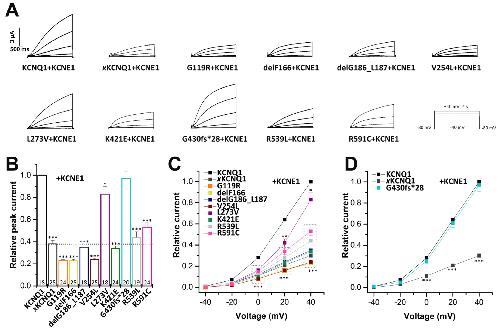
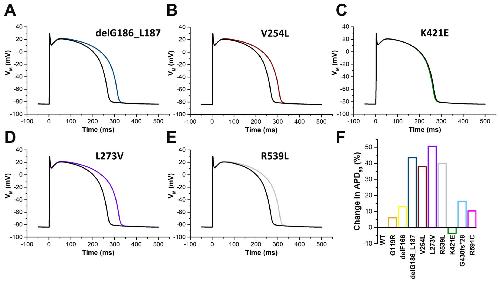
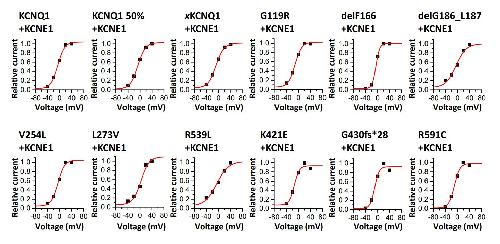
Rinné S,Oertli A,Nagel C,Tomsits P,Jenewein T,Kääb S,Kauferstein S,Loewe A,Beckmann BM,Decher N,
Int J Mol Sci. January 10, 2023; 24(2):1422-0067.
|
| Functional and clinical characterization of a novel homozygous KCNH2 missense variant in the pore region of Kv11.1 leading to a viable but severe long-QT syndrome., Delinière A,Jaupart L,Janin A,Millat G,Boulin T,Andrini O,Chevalier P, Gene. March 1, 2024; 897:1879-0038. |
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}