Designing CRISPR guide RNAs - sample workflow
Designing CRISPR-Cas9 gRNAs targeting Xenopus sequences:
This is a suggested workflow for designing guide RNAs (gRNAs) utilizing three online tools:
1. CRISPRScan (www.crisprscan.org) visualizes where predicted gRNAs are on the gene and the user can choose high scoring sequences. CRISPRscan tracks are also hosted on the Xenbase JBrowse for X. laevis v9.2 and X. tropicalis v10.0 genomes.
2. inDelphi (https://www.crisprindelphi.design/batch) allows the user to check their selected sequences to see if they have a high chance of producing a frameshift insertion/deletion (the desired outcome) versus an in-frame insertion/deletion (not ideal).
3. CRISPOR (http://crispor.tefor.net) can be used to design genotyping primers around the gRNA target.
Continue below to learn more details on using these tools to design gRNA sequences for CRISPR-Cas9 targeted mutations of your gene of interest.
__________________________________________________________________________
Rationale:
Ideally, you want to aim for gRNA sequences that have:
A) a high CRISPRScan score (>70%)
B) high predicted frameshift frequency (>70%)
C) straightforward genotyping primers (no predicted off targets, same set of primers for multiple targets)
Sometimes these conditions are not possible for one exon. Check if the next exon up or down has better scoring candidates, or target another exon necessary for gene function.
You might notice especially in the CRISPOR browser that there are different methods of scoring gRNA efficiency. This depends on the species with which you are working.
For Xenopus, we are interested in the “Moreno-Mateos” score, the CRISPRScan score. The laboratory that developed CRISPRscan predicted gRNA efficiency using this score in Zebrafish which translates well to Xenopus (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4589495/).
For gene knockout/loss of function gRNAs:
- The gRNA sequence should target as early in the gene as possible (exons 1 or 2), producing a frameshift insertion/deletion, aiming to create a premature stop codon resulting in the gene product truncation of the translated gene.
- In the browser, check to see if the gene is on the forward or reverse strand of DNA. If forward, the introns will have arrows pointing right “>->->->”, and the first exon will be the left most box. If reverse, the introns will have arrows pointing left “<-<-<-<-<” and the first exon will be the right most box. Be sure of the directionality so you do not accidentally design gRNAs for the last exon instead of the first!
For patient-based mutation gRNAs:
- Find a high scoring gRNA as close to the clinically reported mutation location. If it’s in the same exon or neighboring exons, that should suffice. The aim is to disrupt the same protein domain (which can span over a few coding exons). Ensembl browser (https://useast.ensembl.org/index.html) has a good visualization of protein domains and which exons they cover.
__________________________________________________________________________
A: Using CRISPRScan
A1. Open CRISPRscan browser (www.crisprscan.org) and click on “Browser tracks” tab at the top (Figure 1A). Doing so brings up a separate page with different species. X. laevis and X. tropicalis genomes are available. Clicking those links among the other species opens up Xenbase JBrowse (Figure 1B).
Make note of the Legend (Figure 1D). The color coding specifies
High CRISPRscan score - High activity gRNAs, bright green
Low CRISPRscan score, gray-green
gRNAs with potential off-targets, gray
Note: a predicted target may have a Moreno-Mateos score lower than 70, but may highlight as green. This sequence may have high specificity (few off-target hits). The reverse is possible. A predicted target may have a score higher than 70 but highlight as gray. This sequence may have several off-target hits.
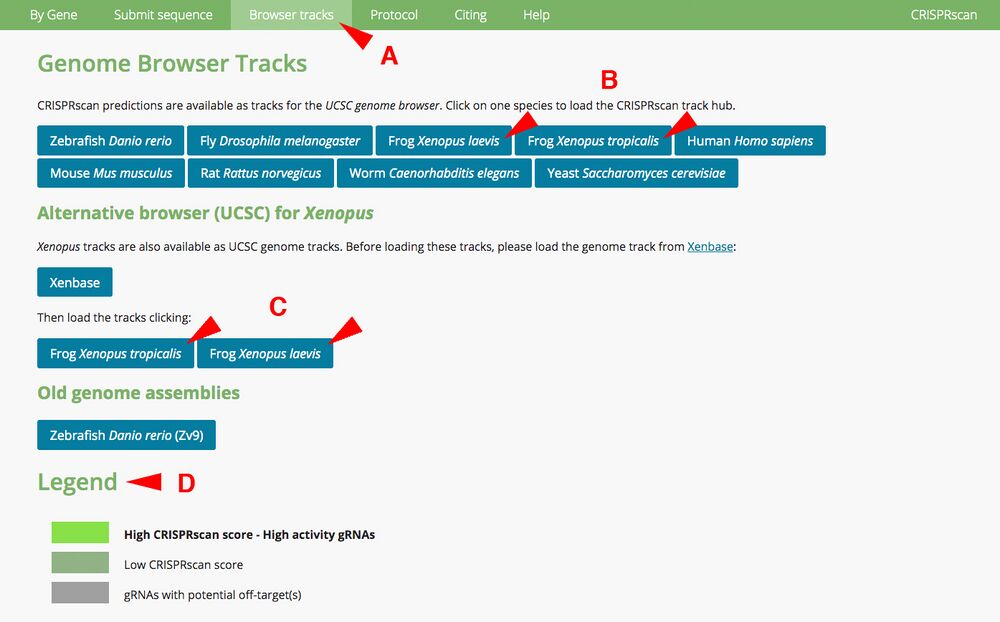
Figure 1.
A2. Within either the Xenbase JBrowse or the UCSC Genome Browser interface, type the name of your gene of interest in the search bar and click “go”. The example used will be for the X. tropicalis foxf1 gene locus.
Load the X. tropicalis foxf1 locus in Xenbase JBrowse (Figure 2A). Access the CRISPRscan tracks by clicking the arrow next to the "CRISPRscan Predictions" and under "Cas9 sgRNAs -> Guide RNAs for coding regions" as an example (Figure 2B).
In this example, we will choose a predicted target in exon 1 of the X. tropicalis foxf1 gene locus.
Mouseover on the bottom predicted gRNA (GA18-NGG87) reveals the Moreno-Mateos score of 87 (Figure 2C). Clicking on the gRNA reveals a pop up window with the associated metadata (Figure 2D).
The highest scoring gRNAs will be in bright green, lower scoring will be in darker green/gray or gray colors.
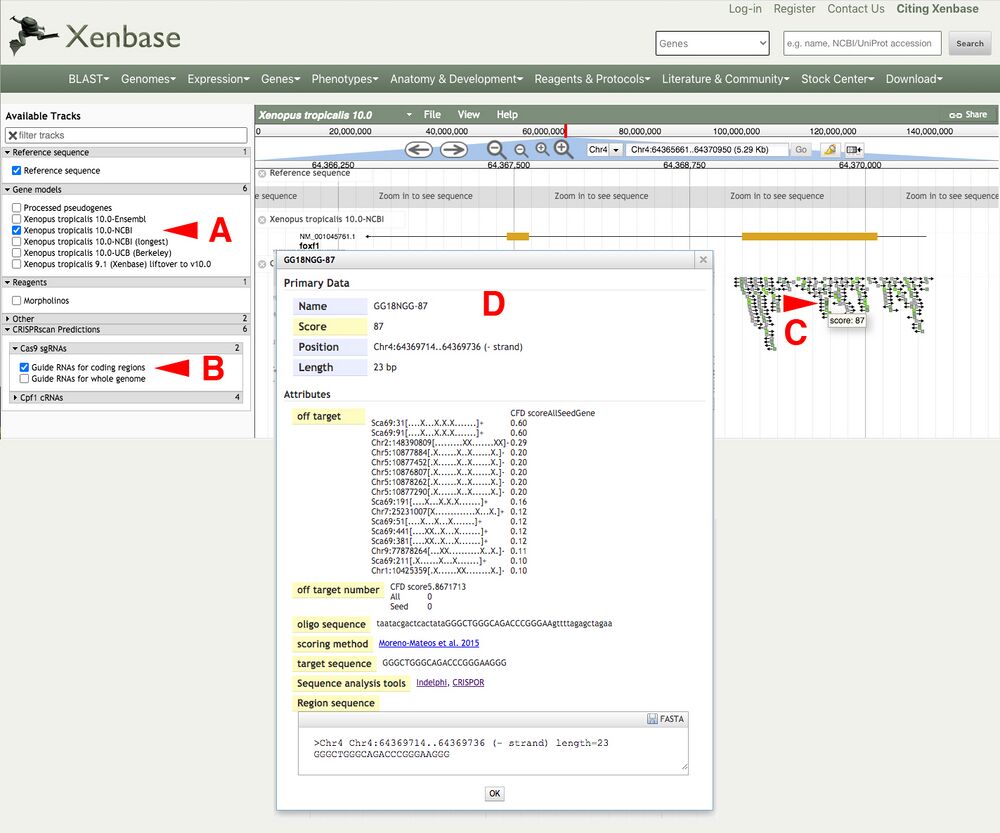
Figure 2.
Zooming in to see the sequence gives higher resolution to see the predicted target gRNA aligned to the genomic sequence (Figure 3, red arrow).
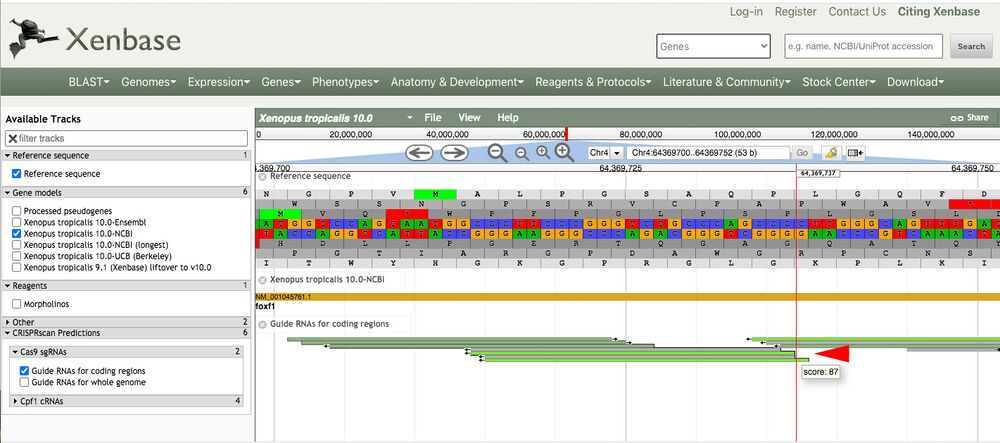
Figure 3.
A3. The predicted gRNA metadata can also be accessed under "Alternative browser (UCSC) for Xenopus, follow the instructions to load the genome track from Xenbase. The foxf1 locus is shown as an example.
link for instructions to load track hub: http://ftp.xenbaseturbofrog.org/pub/sequence_information/RIMLS-SVH/ChIP-seq_Feb2015/trackHub_instructions.html
The search engine may produce a page that has multiple search results. Click on the top selection under “NCBI RefSeq genes”.
A4. Find the first exons and zoom in on the browser by clicking and dragging the region you want to which you want to zoom in.
The highest scoring gRNAs will be in bright green, lower scoring will be in darker green/gray or gray colors.
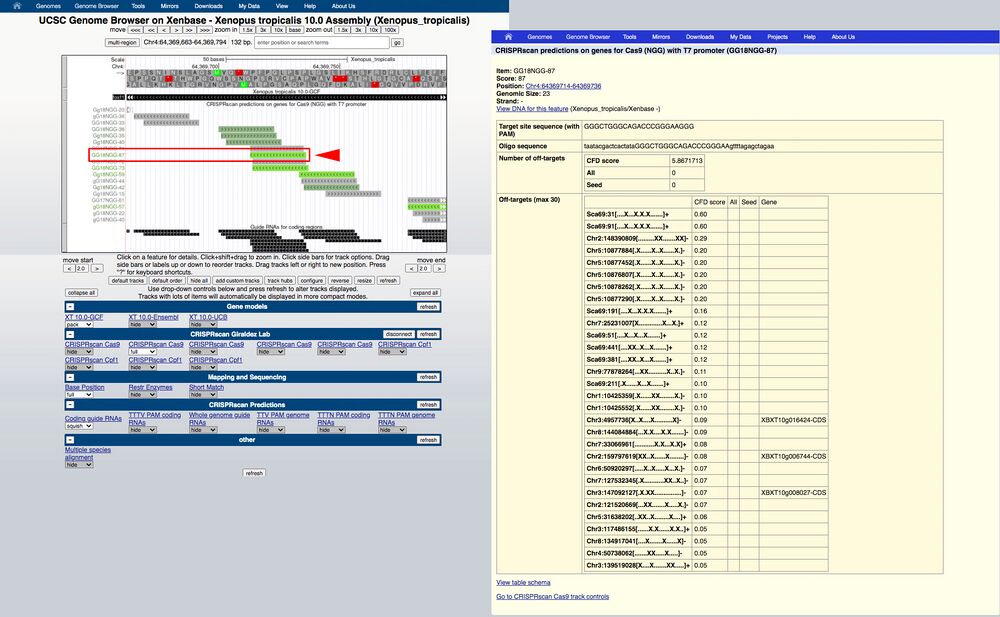
Figure 4.
A5. Click on the green box to get the gRNA sequence and view the associated metadata. We are interested in the “Target site sequence (with PAM)”
A6. Document the chosen gRNA sequence and make sure to put a space between the sequence and the last three letters (the PAM site), to order gRNAs from a company do not include the PAM sequence (separated out with a space):
e.g. the above sequence is written as: GGGCTGGGCAGACCCGGGAA GGG
This process can be repeated within the same segment of DNA should there be two or more high-scoring predicted gRNA targets within the same exon contained within the same segment of genomic DNA. For simplicity sake, we will continue with one set of results for one target.
A7. Once you have your gRNA sequences you can double check their predicited efficiency on inDelphi (continued below in section B).
B: Using inDelphi
B1. In the InDelphi browser (https://www.crisprindelphi.design/batch), go to “BATCH MODE”. Paste a minimum 70bp sequence of the Xenopus genome into this box to get gRNA scores. Go back to the UCSC genome browser to acquire the DNA sequence.
B2. To acquire the DNA sequence in the UCSC genome browser, zoom in or out to a small region around where the gRNA target sequence is located to capture enough sequence for analysis (at least 70bp). Center your selected gRNA target in the browser window. Then in the top blue toolbar, hover the cursor over “View”, then click “DNA”, then click the “get DNA” button.
The FASTA DNA sequence taken from the UCSC browser from above is:
>hub_47641_Xenopus_tropicalis_dna range=Chr4:64369541-64369936 5'pad=0 3'pad=0 strand=+ repeatMasking=none
CACTTGCCCCTTGAAAGCTGTAAGTCTCTGGTATGTGGTTGAAGCCCAGG
CCATTCATCATGCTATACATGGGTTTGAGAGCTTGGCATTTCCTCCTGAA
GCCTCTTGGTCTTCTTCTAAAGGAGCCCTCCTCAAACATAAACTCACTGG
CCGGGTCAATGGTCCAGTAATGGCCCTTCCCGGGTCTGCCCAGCCCCTTG
GGCAGTTTGATAAAGCACTCATTTAGGGACAGGTTGTGCCTGACAGAGTT
TTTCCAGCCCTGGTAGGAGCCTCGGAAAAATGGGAAGCGGCTCTGCAGAA
ACTGGTAGATCTCGCTGAGGGTGAGCCTTTTGGTGGGGGAGCTCTGGATG
GCCATGACTATTAGCGCAATGTACGAGTAAGGCGGCTTTTCTGGCC
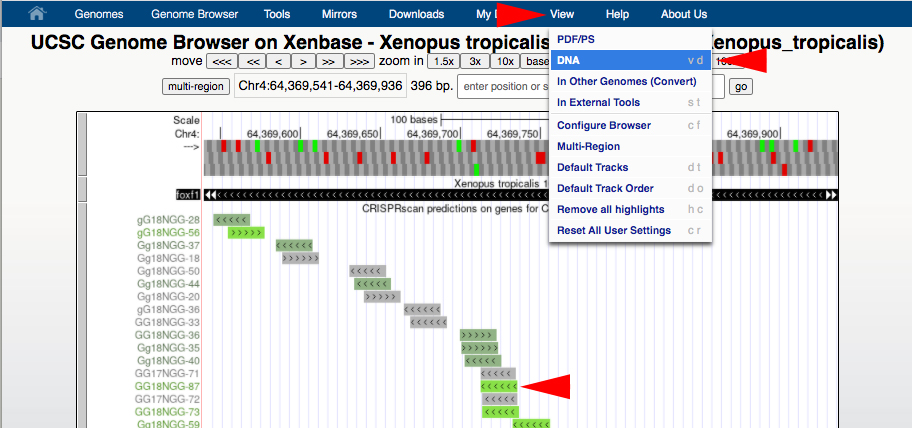
Figure 5.
B3. Copy and paste the DNA sequence into the inDelphi box. It may ask you to “sanitize your sequence” which means just remove any paragraph spaces in the sequence.
link to sanitize DNA sequence: http://www.cellbiol.com/scripts/cleaner/dna_protein_sequence_cleaner.php
B4. Hit “predict repair for X gRNAs” (X will change depending on the number of gRNA sequences the browser finds).
B5. You will see a screen shown in Figure 6. On the left, you want to try to find the sequences from CRISPRScan. Looking for the PAM sequence helps. Sometimes inDelphi can’t find our CRISPRScan sequence, which is ok. Once you find the sequence, click on “Details”. The readout is shown in Figure 6.
B6. Here we are interested in the summary data on the page as seen in Figure 7. Ideally, we want a gRNA sequence with high precision (no off target effects), high microhomology strength, and high frameshift frequency. If the gRNA has “typical frameshift frequency” that is ok too, just avoid any sequences that have low frequencies.
Tip: use the Find function in the browser to find your target sequence. Hit Command-F on Mac or Control-F in Windows and paste in your gRNA sequence WITHOUT the PAM to find the sequence on the page.
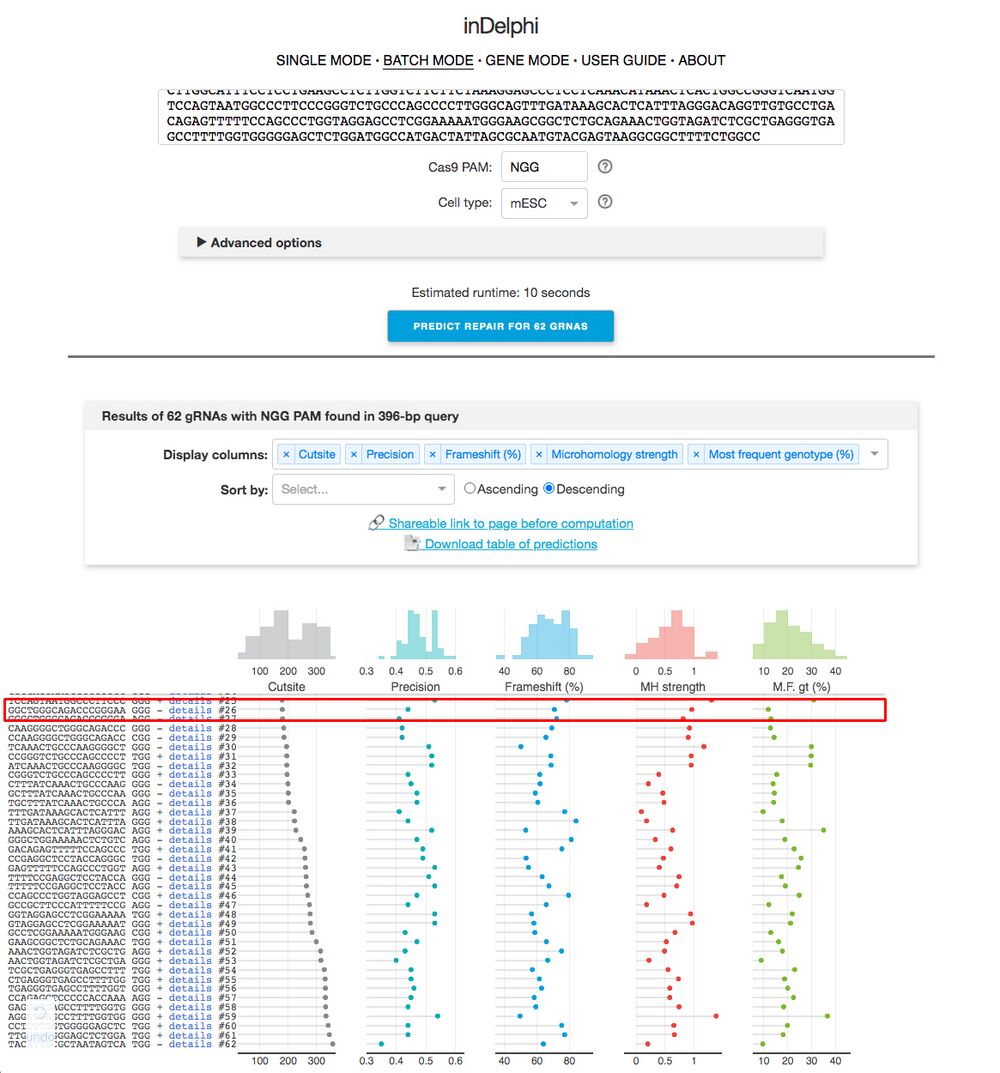
Figure 6.
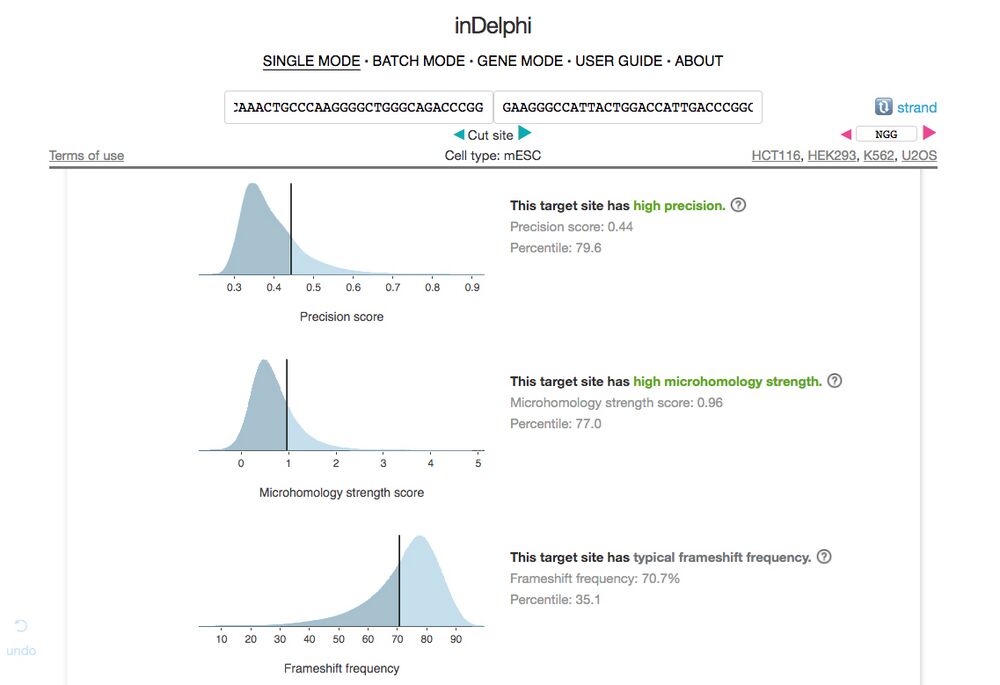
Figure 7.
B7. If gRNA sequences have good scores from inDelphi, move on to designing primers for genotyping (section C below). If not, you can go back to CRISPRscan and try to look for other target sequences that have a good balance between CRISPRScan scores and inDelphi scores (sections A and B).
C: Using CRISPOR for genotyping primers
C1. Open CRISPOR (http://crispor.tefor.net) and copy the DNA sequences containing your gRNAs from the UCSC browser into the box under CRISPOR “Step 1”. For “Step 2”, If using X. tropicalis make sure you choose the Xenopus tropicalis – Western Clawed Frog genome. You can leave “Step 3” as Cas9 PAM, 20bp-NGG as processed thus far (Figure 8, red arrows).
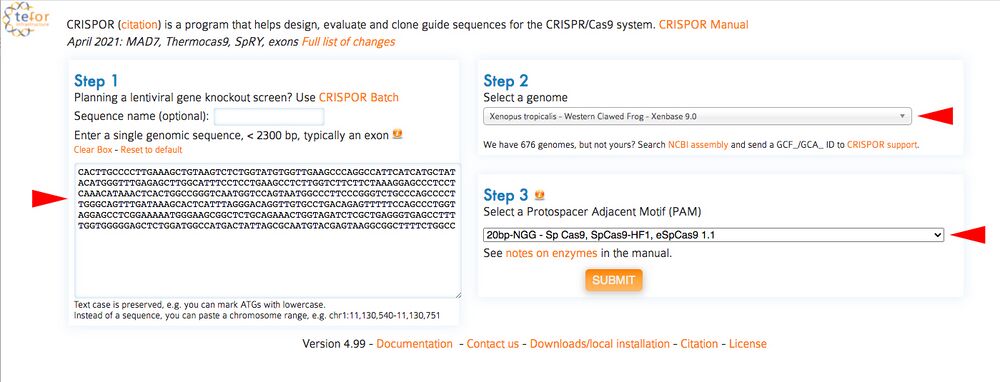
Figure 8.
C2. The aligned targets aligned to the DNA sequence is in the top window. Scroll left and right in the top window to see the targets. The search output below will have a table of possible gRNA sequences on the section of DNA. Find your high scoring gRNA sequences from CRISPRScan and click “Cloning/PCR primers” underneath the sequence.
Tip: as above, use the Find function in the browser to find your target sequence. Hit Command-F on Mac or Control-F in Windows and paste in your gRNA sequence WITHOUT the PAM to find the sequence among many the results on the page.
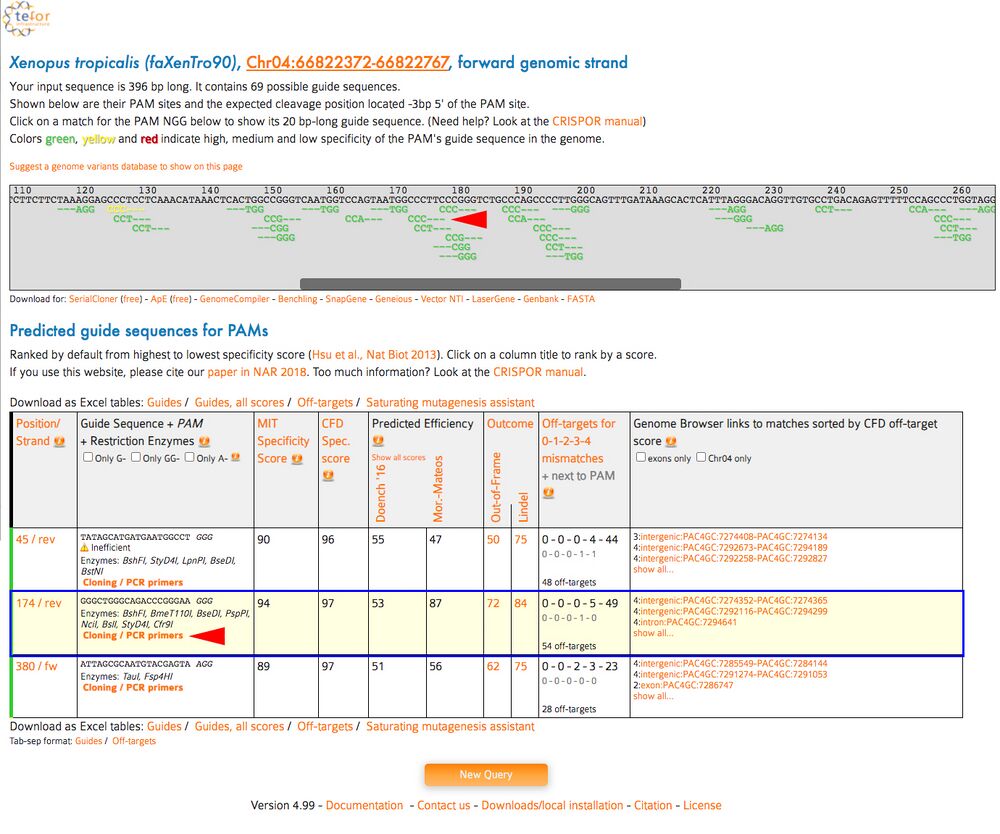
Figure 9.
C3. This will open up a new window with a lot of information. The key section we are interested in is “PCR to amplify the on-target site”. The goal is to have the gRNA sequence (highlighted in yellow) close to the middle of the amplicon from the two recommended primer sequences (underlined). If the yellow sequence is too close to one of the two primers, you can redesign the primers by changing the maximum amplicon length (e.g. set it to 600bp – for Sanger reads) or Primer Tm. Centering the gRNA sequence point when you acquire DNA sequence from the UCSC browser helps in generating the desired amplicon (see section B2 above).
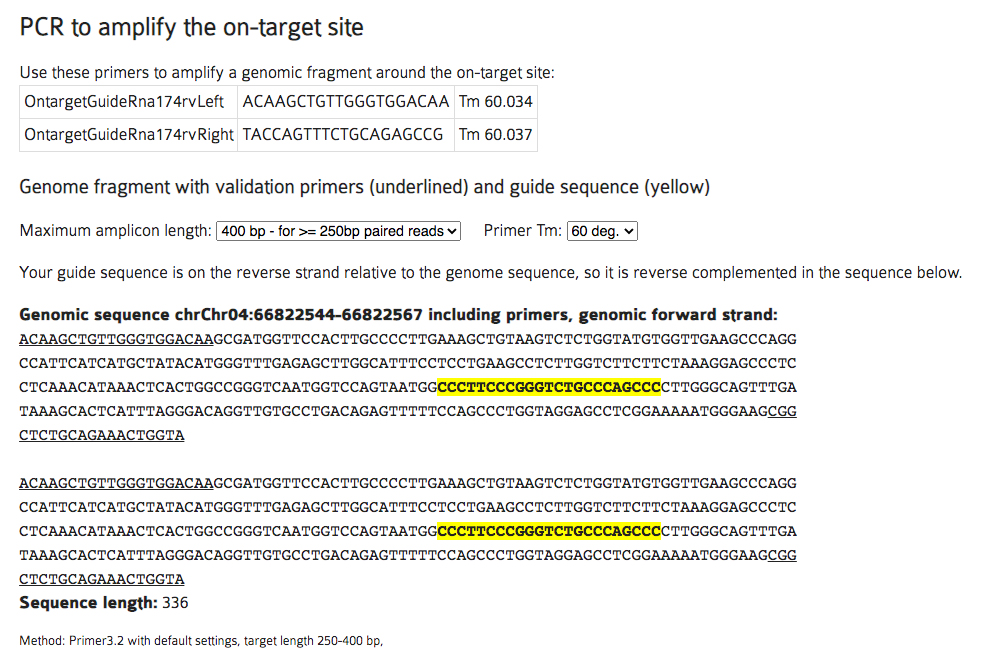
Figure 10.
C4. Double check that the chosen primers will work for both gRNA sequences. To do this, go back into UCSC genome browser and click the “Tools/BLAT” tab. Copy and paste the gRNA sequence, the forward, and the reverse primer into the box and align them all on the genome at the same time.
To do this make sure you separate the sequences by lines starting with ‘>’ e.g. format this way:
>T1
gRNA sequence here
>F
forward primer sequence here
>R
reverse primer sequence here
Based on the gRNA example thus far and the suggested primer sequences from CRISPOR:
>T1
GGGCTGGGCAGACCCGGGAAGGG
>F
ACAAGCTGTTGGGTGGACAA
>R
TACCAGTTTCTGCAGAGCCG
C5. Submitting will take you to a summary page of where all of the sequences hit in the genome. Click on “browser” to the left of any of the sequences to be taken back to the genome browser view. Zoom out so you can see all sequences in the browser, and double check gRNA sequences are nested in the middle of the F/R sequences.
C6. Make sure all of the sequences are clearly documented. The BLAT search is also a final check for specificity of the gRNA sequences and primers. Oligos can then be ordered.
C7. Alternatively, each sequence can be aligned using Xenbase BLAST to check for specificity.
Last Updated: 2021-06-22